Water is the most abundant cellular constituent and therefore important for preserving cellular ultra-structure. Currently the only way to fix cellular constituents without introducing significant structural alterations is by cryo-fixation. There are currently two common methods employed; plunge freezing and high pressure freezing.
Cryo-fixation has two distinct advantages over chemical fixation. It is achieved within milliseconds and it ensures simultaneous immobilization of all macromolecular components. Many protein networks are very labile and fall apart with the slightest osmotic or temperature change and these unwanted effects are minimized during cryo-fixation. These techniques allow the study of biological samples with improved ultra-structural preservation, and can facilitate the study of dynamic processes. Currently, the only method to vitrify thicker samples (up to 200 µm) is by High Pressure Freezing.
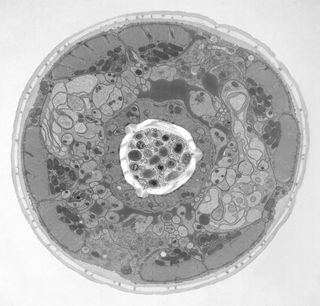

Mouse embryonic fibroblast grown on sapphire disc (courtesy of Verkade P, University of Bristol, UK).
Successful cryo-fixation (vitrification) is the transformation of water from a liquid to an amorphous state without inducing the nucleation of ice crystals. The nucleation of ice crystals is temperature- and pressure-dependent (see diagram below). Crystallisation also depends on the cooling rates as freezing is a time dependant process. The cooling rates depends on the thermal properties of water, the sample thickness and the heat extraction flow at the surface of the specimen.

The idea of freezing biological samples under pressure was first introduced by Moor and Riehle [1]. The High Pressure Freezing device became commercially available in 1985. All High Pressure Freezing machines available currently, despite having different design, deliver synchronized pressurization and cooling of the sample within 20 ms in a highly reproducible manner.
The diagram by Kanno et al. [2] shows the states of water depending on pressure and temperature. At a pressure of 2,045 bar the melting point of water is lowered to 251 K and the temperature for homogenous nucleation is reduced to 181 K.
Based on the principle of Self Pressurized Rapid Freezing introduced by Leunissen and Yi [3] a new technology has been developed by Leica Microsystems. It uses the tendency for water inside a sealed specimen carrier to expand upon cooling, thereby generating pressure intrinsically instead of using an external hydraulic system. This pressure is likely to be the result of crystalline and low density ice formation within the sealed specimen carrier. To achieve pressure (2,010 bar) where the melting point of ice is lowered to 251 K (Kanno et al. [2]) 60 % of the water inside the specimen carrier needs to be converted to low density ice.
Low density ice formation causes a volume expansion relative to liquid water. Numerical simulations show that freezing along the tube walls proceeds freezing in the center, producing a strongly curved ice front. This effect is prominent when the immersion speed is the highest. The separation between regions containing low density ice and well-frozen or vitrified parts is considered to be best when the ice front is as flat as possible. This can be achieved by alteration of the freezing parameters for each specific case within the Leica EM SPF program interface. During the immersion movement, the ice formation front inside the tube is always above the level of the cryogen surface. The distance of this ice front depends on the immersion speed.

Fig. 1: Overview images of PtK2 cells grown on sapphire discs: The finder grid located close above the cells for retracing the sample.

Fig. 2: DIC and fluorescent images: When a cell of interest was located DIC and fluoresenct images (internalized EGF-QD655) were made. The overlay (right image) shows the location of the EGF-QD655 containing structures in the cell. The boxed area is the area of interest within the cell.

Fig. 3: Fluorescent quantum dots inside the cell of interest: The structures containing the fluorescent quantum dots inside the cell of interest were followed live, making a movie sequence with images taken every second. Still images, showing images 5 seconds apart, from this movie sequence are shown. The last image was the last image of the sequence (hence time = 0 s). At this moment the rapid loader was transferred from the stage insert into the RTS and the sample was frozen.

Fig. 4: Electron micrograph of the cell of interest (left), zoom into structure of interest (right): Electron micrograph of the cell of interest (left). The boxed area is the area that contains the structure of interest and a zoom into the structure of interest is shown on the right. Arrows indicate quantum dots that can be identified inside the structure. Compare the C-shape of this structure with the last image of Figure 3, and a comparable C-shape can be observed. Note that the electron micrograph is from a 70 nm section while the fluorescent image is approximately 1 μm thick.
Courtesy of Verkade P, University of Bristol, UK.
Related instruments: Leica EM PACT2 with Leica EM RTS
The sections are of yeast frozen with a Leica HPM100 in the copper tube system, the cell paste was mixed with a pH 6.5 MES/dextran buffer so that a final MES concentration of 50 mM and a dextran concentration of 20 % was achieved.
The samples were sectioned on a Leica EM UC7/EM FC7 with Micromanipulator at –140 °C and the section thickness was set to 50 nm. The sections were attached to Agar lacey grids.
The sections were imaged using a Tecnai Polara 300KeV (FEI, The Netherlands) microscope fitted with a 4 K Gatan CCD camera. The magnification for the sections was 23 K, with a defocus of –6 um for the tomogram and –8 um for the projection image, and the diffraction was done with a camera length of 930 mm. The image in the left part of the panel is an average of the central 10 slices of a reconstruction done with the IMOD package (Kremer et. al. [4]) image processing software from a tomogram collected using the FEI software.

Courtesy of O'Driscoll J, Clare DK and Saibil H, prepared at the Department of Cryostallography, Birkbeck, University of London.
Related instruments: Leica EM HPM100

Fig. 1: Pyramidal cell: Synaptic connection between a bouton (P-face) and a dendritic spine (E-face) of a pyramidal cell (courtesy of Kulik A, Institute of Anatomy and Cell Biology, University of Freiburg, Germany); related instrument: Leica EM HPM100.

Fig. 2: Higher magnification of inclusion of cell in Figure 1. (C), Chlamydia pneumoniae cells; (G), Glycogen granules; (Go), Golgi; (M), Mitochondrium; (Mt), Microtubules; (R), Ribosomes; (V), Vesicles. Scale bar 500 nm (courtesy of Kaech A, Center for Microscopy and Image Analysis, University of Zurich, Switzerland); related instrument: Leica EM HPM100.
 |
 |
| Fig. 1: CEMOVIS of Saccharomyces cerevisiae (courtesy of Al-Amaudi A, DZNE, Bonn); related instrument: Leica EM SPF. | Fig. 2: Freeze substitution of Pseudomonas deceptionensis (courtesy of Lopez-Iglesias C, Delagado L and Mercade E, CC i-University Barcelona, Science Park); related instrument: Leica EM SPF. |

Fig. 3: Freeze substitution of Caenorhabditis elegans (courtesy of the Delaware Biotechnology Institute Bio-Imaging Center [Modla S, Jacobs S, Caplan J and Czymmek K] and University of Pennsylvania [Tanis J]).

Fig. 4: Freeze substitution of Lingulodinium polyedra (extrusomes) (courtesy Lindemann E, Fraunhofer IG B [[Functional Genomics], Stuttgart]); related instrument: Leica EM SPF.








